

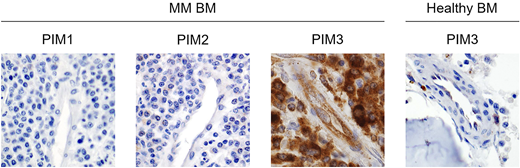
The development and progression of multiple myeloma (MM) depend on the formation and perpetual evolution of an immunosuppressive and hypervascular bone marrow microenvironment. MM undergoes an angiogenic switch during its early progression stages and initiates the secretion of proangiogenic proteins, such as VEGFA and Galectin-1. Following their engagement with the VEGF receptor 2 on the surface of the endothelium, quiescent endothelial cells (ECs) rapidly switch to an activated state, thus gaining the ability to create sprouts, migrate and proliferate. However, chronic angiogenic stimulation results in the formation of a dense and leaky network of pathological vessels, which in the case of MM also serves as a major source of prosurvival paracrine signals. Since PIM kinases are known modulators of cytokine signaling, owing to their ability to activate NFκB, JAK/STAT and mTOR pathways, we analyzed the expression pattern of PIM1, PIM2 and PIM3 in multiple myeloma bone marrow samples using immunohistochemistry. We found that both MM cells as well as myeloma-associated ECs exhibit a significantly higher PIM3 expression than their normal bone marrow counterparts. Since the role of PIM kinases in the vascular compartment of the tumor microenvironment is currently unknown, we decided to explore the proangiogenic functions of PIM kinases using in vitro MM and EC model cell lines.
3 MM cell lines (RPMI 8226, MM1.s, U266), immortalized bone marrow ECs (HBMEC-60) and human umbilical vein ECs (HUVECs) were used for the experiments. Primary MM cells were obtained from MACS-separated bone marrow aspirates. Chemical blockade of PIM kinase activity was achieved using the pan-PIM inhibitor SEL24/MEN1703. The compound decreased the viability of MM cell lines with IC50 in the submicromolar range, induced G2 cell cycle arrest and apoptosis. Moreover, SEL24/MEN1703 induced apoptosis in primary MM cells, even when cocultured with the CD138- bone marrow fraction. PIM inhibitor treatment inhibited the phosphorylation of mTOR substrates S6 and 4EBP1, STAT3/5, as well as RelA/p65. Consequently, we observed markedly decreased VEGFA and Gal-1 levels in SEL24/MEN1703-treated MM cells. When cultured together, separated by a permeable transwell membrane, both RPMI 8226 cells, as well as ECs, exhibited a 2-fold increase in proliferation rate. This effect was completely blocked by a 2-day treatment with a PIM inhibitor.
Exposure of ECs to recombinant VEGFA (10ng/ul) or MM supernatant resulted in an increase in VEGFR2 Y1175 phosphorylation level and induction of PIM3 expression. Increased MYC activity is a hallmark of VEGF-dependent endothelial activation and is necessary to support the creation of new vessels. Since the PIM3 promoter region contains putative MYC-binding sites (E-boxes), we checked if PIM3 induction depends on MYC in ECs. MYC silencing using siRNA resulted in an 88% lower PIM3 expression than the non-targeting siRNA. One of MYC's main tasks during angiogenesis is the stimulation of cellular ATP synthesis to meet the energy demands created by the dynamic remodeling of the actin cytoskeleton. Surprisingly, PIM inhibition decreased the total ATP content in ECs by 25%, thus disrupting the energetic homeostasis, as evidenced by a 9.6-fold increase in phosphorylated AMPK T172 levels. Furthermore, SEL24/MEN1703-treated ECs were depleted of higher-order actin structures necessary for efficient angiogenesis, such as actin stress fibers, membrane ruffles and lamellipodia. In consequence, PIM kinase inhibition decreased proliferation, migration and formation of new vessel-like structures in Matrigel by ECs.
Collectively, our data demonstrate that PIM inhibition induces MM cell death and abolishes important tumor cell-ECs interactions. In addition, we show that PIM3 is overexpressed in MM tumor endothelial cells and PIM inhibition disrupts the activation state in in vitro cultured ECs. Hence, targeting PIM kinases may represent an efficient approach to induce tumor cell death and to block angiogenesis in MM. RNA-sequencing studies on the downstream effectors of PIM3 are currently ongoing in order to unravel the molecular mechanism behind the observed effects.
Brzózka:Ryvu Therapeutics: Current Employment. Rzymski:Ryvu Therapeutics: Current Employment. Tomirotti:Menarini Ricerche: Current Employment. Lech-Marańda:Roche, Novartis, Takeda, Janssen-Cilag, Amgen, Gilead, AbbVie, Sanofi: Consultancy; Roche, Amgen, Gilead: Speakers Bureau. Juszczynski:Ryvu Therapeutics: Other: member of advisory board.

